




Related news


Oxidative Stress Molecular Mechanisms and Signaling Pathways: What Really Happens Inside Cells
Table of Contents
Oxidative stress represents a core research hotspot across cell biology, toxicology, pharmacology, geroscience, oncology and neurodegenerative disease modeling. Its core definition sounds straightforward: intracellular reactive oxygen species (ROS) accumulate to levels exceeding the cell’s intrinsic antioxidant clearance capacity and disrupt redox homeostasis. However, practical cell culture experiments reveal far more complex regulatory logic. Mild, transient ROS bursts serve as vital secondary messengers to mediate physiological signaling, while sustained, high-concentration ROS overload triggers irreversible damage to DNA, proteins and membrane lipids, sequentially driving mitochondrial dysfunction, inflammatory cascades, apoptotic cell death and diverse cellular stress responses.
For labs conducting ROS fluorescent detection, antioxidant efficacy screening and inflammatory stress pathway research, quantifying the magnitude, duration and subcellular localization of ROS elevation matters far more than simply confirming a generic ROS rise. Multiple confounding variables can drastically skew experimental readouts, including cell line origin, drug incubation duration, fluorescent probe loading efficiency, reagent grade, cold-chain storage protocols and post-treatment sample processing. Solarbio delivers comprehensive life science research supplies covering biochemical reagents, small-molecule bioactive compounds, primary/secondary antibodies, ELISA detection kits and cell culture consumables, fully catering to molecular, immunological and biochemical pathway research demands and helping researchers minimize experimental bias.
What Is Oxidative Stress?
Oxidative stress refers to a disrupted intracellular redox steady state, caused either by excessive ROS generation or impaired antioxidant detoxification capacity. While sustained high levels of ROS exert severe cytotoxicity, low-dose, transient ROS pools function as essential signaling mediators to modulate immune activation, cell proliferation and tissue developmental processes. Oxidative stress onset is triggered by three key conditions: excess bulk ROS accumulation, prolonged ROS overload within redox-sensitive subcellular compartments (mitochondria and nucleus), and long-term unbalanced redox status, all of which initiate downstream cytotoxic cascades.
Main ROS Types
Diverse reactive oxygen species exist within eukaryotic cells; the three most extensively studied subtypes are superoxide anion radical (O2–•), hydrogen peroxide (H2O2), and hydroxyl radical (•OH). Superoxide anion radicals are predominantly generated alongside mitochondrial electron transport chains. H2O2 exhibits superior chemical stability and high membrane permeability, enabling it to travel across organelle boundaries and act as both a redox signaling messenger and cellular stress sensor. In stark contrast, hydroxyl radicals possess extreme chemical reactivity and instantly oxidize nearby DNA, proteins and membrane lipids upon formation.
Simply detecting elevated total ROS levels cannot support rigorous oxidative stress research. ROS upregulation may stem from distinct cellular stimuli with variable durations, and downstream signaling cascades exhibit staggered activation timelines. To acquire comprehensive, reproducible experimental data, researchers need a complete research toolkit covering stable cell stress models, multi-index biochemical detection kits and pathway-specific analysis reagents — all of which are fully supplied by Solarbio to simplify your multi-dimensional redox profiling.
ROS Sources Inside and Outside the Cell
Mitochondria constitute the dominant endogenous ROS source. During mitochondrial oxidative phosphorylation, electron leakage from electron transport chain Complex I and Complex III enables free electrons to react with molecular oxygen to generate superoxide radicals. The endoplasmic reticulum also generates ROS under unfolded protein response stress. Peroxisomal fatty acid β-oxidation and NADPH oxidase activation (the major ROS source in immune and inflammatory cell models) further contribute to intracellular ROS pools.
Exogenous stressors can drastically boost cellular ROS abundance: ultraviolet and ionizing radiation directly induce radical formation; heavy metals suppress antioxidant enzyme activity and disrupt mitochondrial function; tobacco-related toxicants, excessive ethanol, airborne particulate matter and chemotherapeutic agents collectively break redox homeostasis. Notably, routine cell culture conditions can alter basal ROS levels and introduce experimental bias, including basal medium formulation, fetal bovine serum batch, cell passage count and seeding density. These easily overlooked variables frequently lead to substantial discrepancies between parallel groups. Solarbio provides standardized cell culture media, low-endotoxin serum and uniform cell dissociation reagents to minimize such experimental interference.
How Does ROS Damage Cells?
Normally, ROS functions as a signal molecule. However, at a certain level it can start to damage vital cellular components.
DNA Damage
Highly reactive hydroxyl radicals trigger oxidative modification of DNA guanine bases to form 8-OHdG, the canonical biomarker of oxidative DNA injury. These radicals also induce single- and double-strand DNA breaks and irreversible DNA-protein crosslinking. Transient DNA lesions trigger temporary cell cycle arrest and activate endogenous DNA repair pathways, yet persistent or recurrent oxidative damage drives genomic instability, cellular senescence and malignant transformation.
Protein Oxidation
ROS directly oxidize amino acid side chains within polypeptides: cysteine sulfhydryl groups undergo reversible oxidation, while methionine methylene groups form carbon-carbon double bonds after oxidation. The oxidation intensity produces diametrically opposed functional outcomes: mild cysteine oxidation acts as a reversible redox switch for signal transduction, whereas robust oxidative modification triggers irreversible protein misfolding, functional inactivation and insoluble aggregate formation. This pathway is central to neurodegeneration research: sustained ROS overload impairs damaged protein clearance systems and accelerates β-amyloid plaque aggregation, a hallmark pathological feature of Alzheimer’s disease.
Lipid Peroxidation
Plasma and organelle membranes are enriched in polyunsaturated fatty acids, which are highly susceptible to ROS oxidative attack. Lipid peroxidation generates stable end products malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE), the most commonly quantified biomarkers of lipid oxidative injury. These toxic aldehydes disrupt membrane integrity, impair mitochondrial function, crosslink with cellular proteins and DNA, and create a positive feedback loop to amplify inflammatory signaling. To obtain robust, multi-dimensional experimental data, most research groups simultaneously detect ROS abundance, lipid peroxidation levels, antioxidant enzyme activity, pro-inflammatory cytokine secretion and cell viability.
Key Signaling Pathways in Oxidative Stress
Oxidative stress does not solely translate into one line of subsequent cellular events. As ROS can cause damage to individual molecules as well as activate or modulate signal transduction pathways, a variety of different cellular responses can be initiated by oxidative stress, which in turn depend on the intensity and duration of oxidative stress as well as on the cellular background.
Nrf2-ARE Pathway: Antioxidant Defense
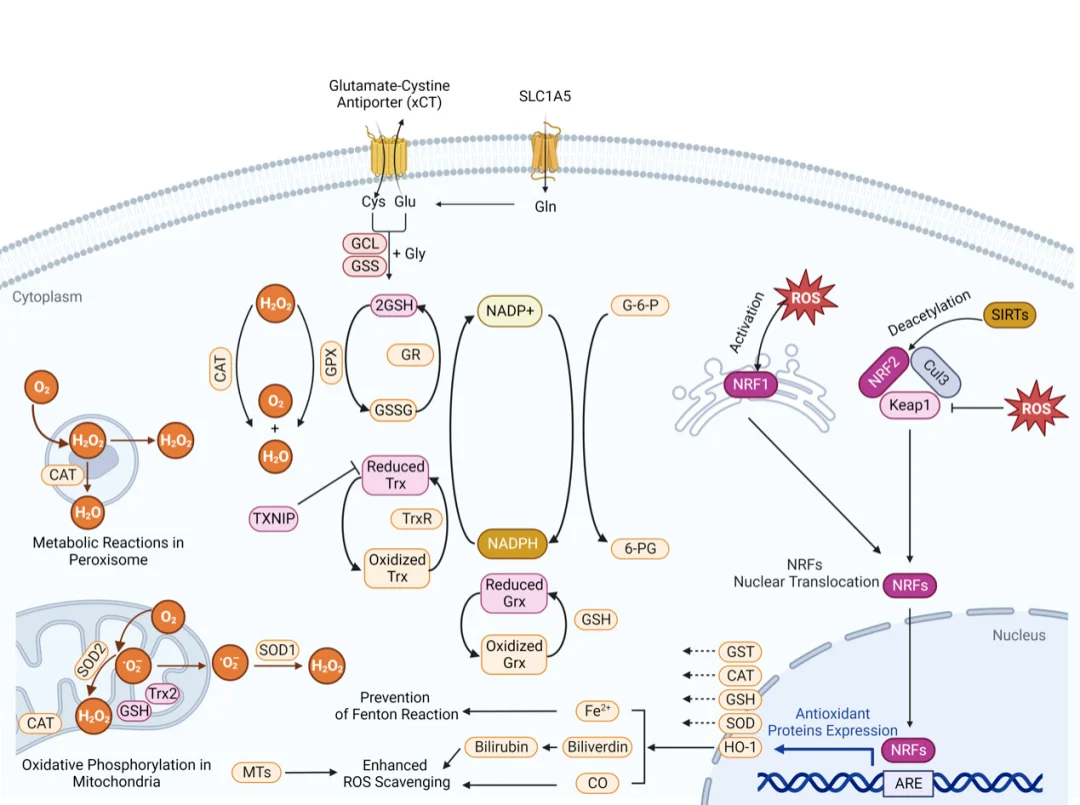
The Nrf2-ARE cascade serves as the master regulatory pathway governing cellular antioxidant defense. Under physiological redox homeostasis, cytoplasmic Nrf2 binds tightly to Keap1 and undergoes continuous ubiquitination and proteasomal degradation. When ROS accumulate, free radicals covalently modify cysteine residues on Keap1, altering its conformation to dissociate Nrf2; meanwhile, activated PI3K/Akt phosphorylates Nrf2 to further accelerate its nuclear translocation. Inside the nucleus, Nrf2 heterodimerizes with small Maf proteins and binds to antioxidant response elements (AREs) on target gene promoters.
This transcriptional program upregulates antioxidant enzymes (SOD, CAT, GSH-Px), glutathione synthetic enzymes and phase II detoxification proteins, collectively enhancing ROS scavenging capacity and cellular damage repair. Extensive preclinical models confirm Nrf2 activation exerts cytoprotective effects in hepatic, pulmonary, neuronal and epithelial tissue injury.
Notably, pathway readouts exhibit asynchronous temporal dynamics: Nrf2 nuclear translocation, downstream target gene mRNA/protein expression, antioxidant enzyme activity and ROS clearance efficiency do not reach peak values simultaneously.
NF-κB Pathway: The Crosstalk Hub Between Oxidative Stress and Inflammation
The NF-κB cascade acts as the critical molecular bridge connecting ROS overload and inflammatory responses. In unstimulated resting cells, NF-κB dimers bind tightly to inhibitory IκB proteins and remain sequestered within the cytoplasm. Elevated ROS activates the IKK kinase complex, which phosphorylates IκB to trigger its ubiquitination and proteasomal degradation. Free NF-κB translocates into the nucleus and drives transcription of pro-inflammatory mediators, including TNF-α, IL-1β and IL-6.
This pathway forms a self-amplifying positive feedback vicious cycle: ROS initiates inflammatory signaling, and activated immune cells secrete additional ROS to further boost NF-κB activity. Persistent cyclic activation drives the progression of chronic inflammatory disorders, including atherosclerosis, rheumatoid arthritis, hepatic oxidative injury and metabolic low-grade inflammation.
MAPK Cascade: Master Regulator of Cell Proliferation, Stress Response and Apoptosis
Mitogen-activated protein kinase (MAPK) cascades are activated by diverse cellular stressors including ROS, comprising three functionally distinct subfamilies: ERK, JNK and p38 MAPK, which operate via sequential MKK-mediated phosphorylation. Low physiological ROS concentrations selectively activate ERK signaling to facilitate cell cycle progression, proliferation and adaptive redox homeostasis. By contrast, excessive ROS accumulation triggers robust JNK and p38 phosphorylation, initiating cellular stress responses, pro-inflammatory gene transcription and apoptotic cell death.
In myocardial ischemia-reperfusion injury models, rapid oxygen reintroduction induces a sharp ROS burst, hyperactivating JNK/p38 signaling to trigger massive cardiomyocyte apoptosis. In oncology research, basal low-level ROS sustains tumor proliferation via ERK activation, while pharmacologically boosted ROS levels sensitize malignant cells to chemo- and radiotherapy. Collectively, both ROS concentration and exposure duration determine the final cellular fate regulated by MAPK signaling.
PI3K/Akt Pathway: Cytoprotective Survival Signaling Under Moderate Redox Stress
The PI3K/Akt cascade represents the primary pro-survival signaling axis under mild oxidative stress. Moderate ROS stimulation activates PI3K to generate membrane-bound PIP3, which recruits cytoplasmic Akt to the plasma membrane for phosphorylation and full activation. Phosphorylated Akt suppresses apoptotic signaling through multiple mechanisms: it phosphorylates and inactivates the pro-apoptotic protein Bad, inhibits caspase cascade activation, and upregulates mTOR-mediated cellular metabolism and proliferation. Additionally, Akt directly phosphorylates Nrf2 to accelerate its nuclear translocation and amplify antioxidant transcriptional responses.
This protective signaling has a critical redox threshold. Once ROS concentrations exceed the tolerable range, PI3K and Akt undergo irreversible oxidative inactivation and proteolytic degradation, shifting cellular signaling dominance toward the mitochondrial intrinsic apoptotic pathway. In Parkinson’s disease neuronal models, the therapeutic efficacy of antioxidant interventions relies heavily on the intact functional status of PI3K/Akt signaling.
Disease Links in Oxidative Stress Research
Oxidative stress acts as a shared pathological driver of numerous human disorders. Rather than merely identifying cell damage phenotypes, dissecting the underlying redox signaling cascades that trigger tissue injury delivers far more translational research value.
Cancer Research
Tumor cells exhibit heterogeneous basal ROS status: some subtypes maintain inherently high ROS, while others upregulate the Nrf2 antioxidant axis to suppress ROS buildup, which confers strong resistance to chemo- and radiotherapy. That said, artificially boosting intracellular ROS to toxic thresholds can successfully induce malignant cell death. For oncology research, multi-dimensional detection systems are essential, including ROS fluorescent probes, drug sensitivity testing kits, small-molecule compound libraries and pathway-specific biomarker antibodies.
Neurodegenerative Disease Research
Mature neurons are post-mitotic cells incapable of regeneration, featuring fragile energy metabolism and extreme vulnerability to oxidative insults. ROS-mediated impairment of mitochondria and protein folding systems will ultimately initiate apoptotic programmed cell death. Parkinson’s disease animal models demonstrate that excess ROS simultaneously suppresses the cytoprotective PI3K/Akt pathway and hyperactivates pro-apoptotic JNK signaling. In Alzheimer’s disease, ROS overload accelerates pathological β-amyloid aggregation and triggers sustained neuroinflammation; targeted activation of Nrf2 can effectively alleviate these neurodegenerative phenotypes.
Cardiovascular and Liver Injury Models
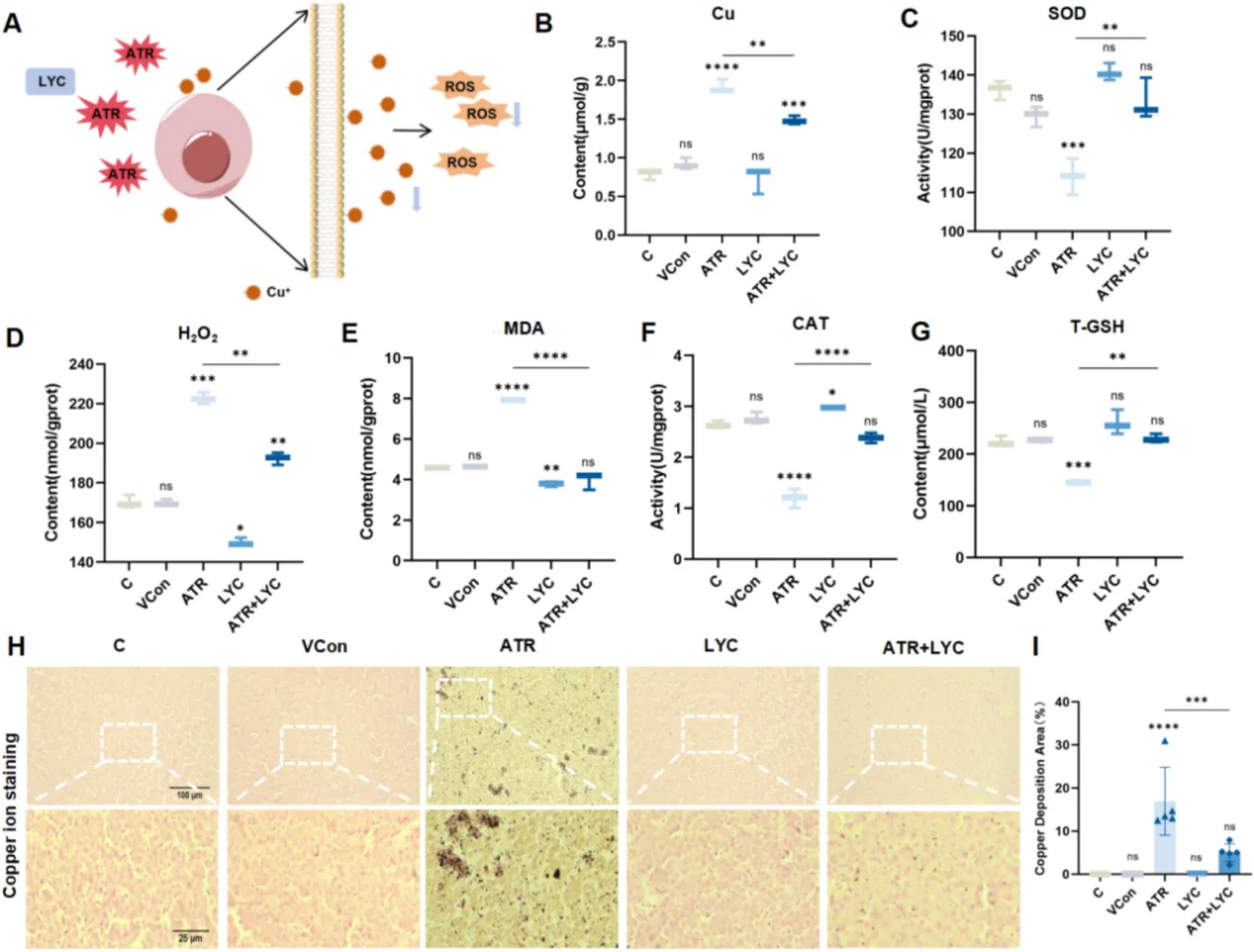
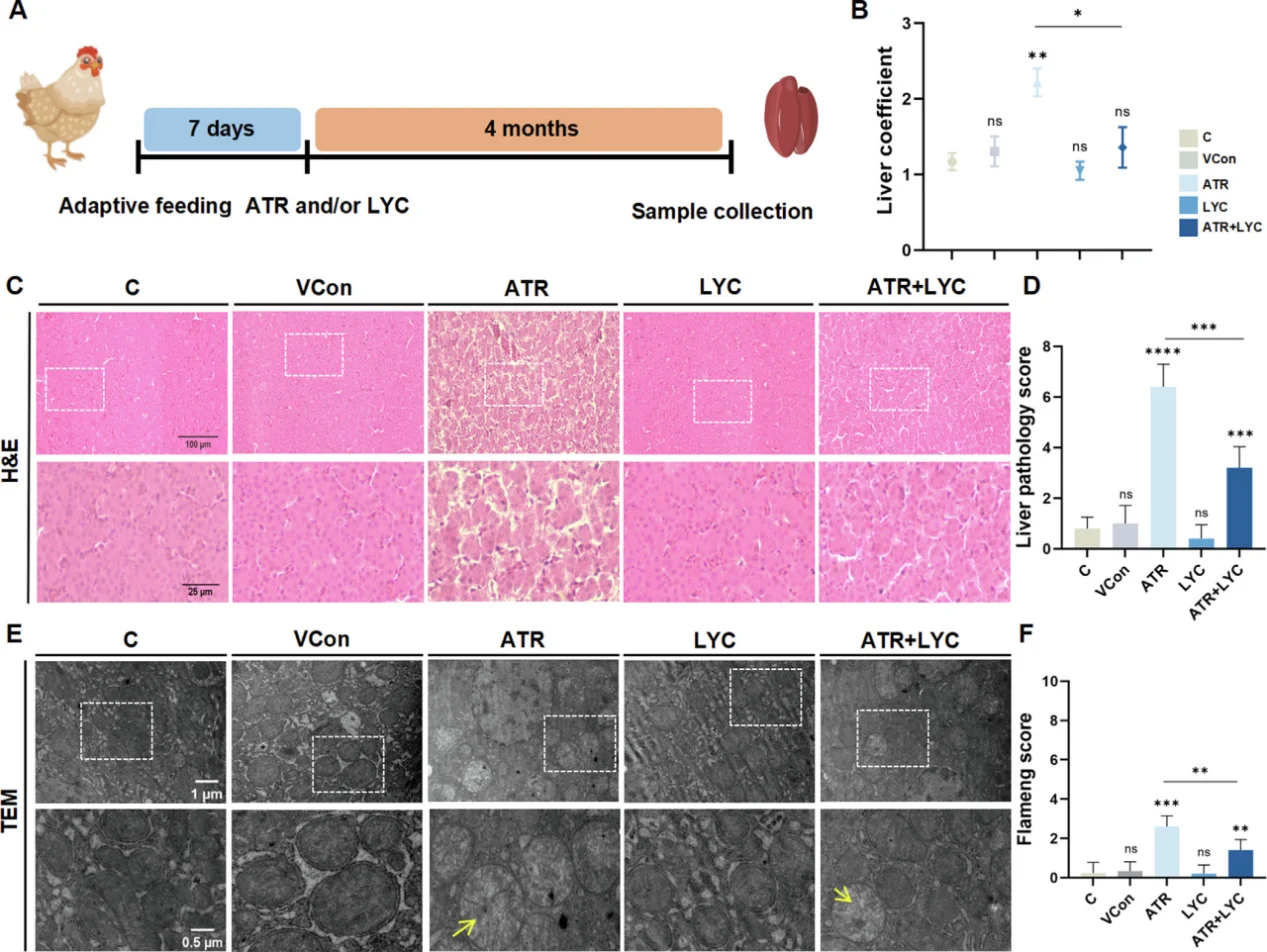
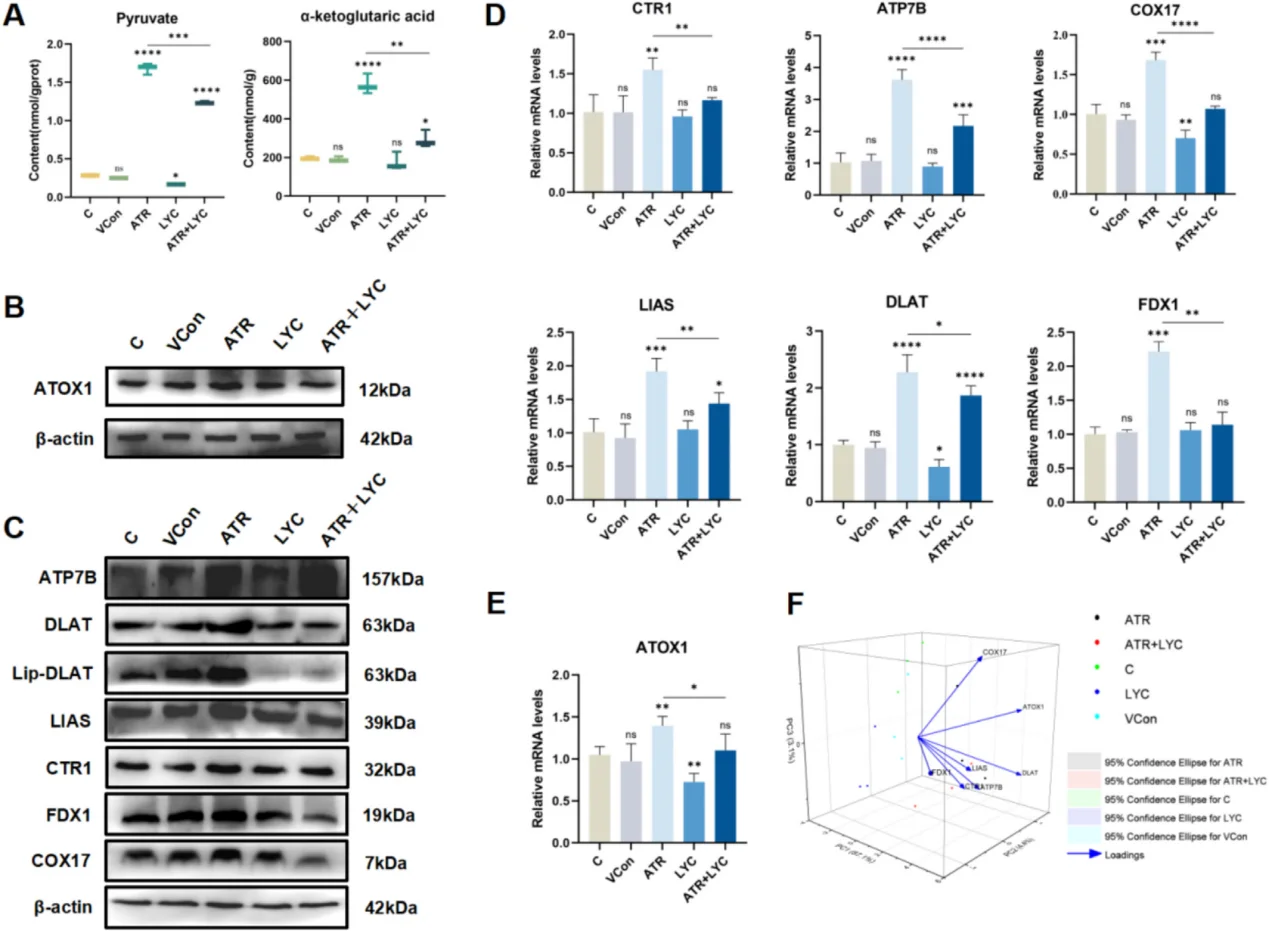
In cardiovascular research, persistent NF-κB activation drives endothelial inflammation and adhesion molecule overexpression, accelerating atherosclerotic plaque formation. A sharp ROS burst arising from myocardial ischemia-reperfusion injury activates JNK/p38 MAPK cascades and triggers massive cardiomyocyte apoptosis. For hepatic toxicology models, oxidative stress triggers mitochondrial dysfunction, massive lipid peroxidation, pro-inflammatory cytokine secretion and cuproptosis. A typical research paradigm employs atrazine to induce liver oxidative damage, with lycopene as the antioxidant intervention agent to rescue tissue lesions.
Product Directions for Oxidative Stress Experiments
For antioxidant rescue experiments, we recommend N Acetyl L Cysteine (Cat. No. IA0050), a classic small-molecule redox modulator for ROS research. NAC replenishes intracellular thiol pools and boosts glutathione-dependent antioxidant systems, enabling researchers to verify whether observed phenotypic changes are driven by ROS-mediated stress or irrelevant off-target effects.
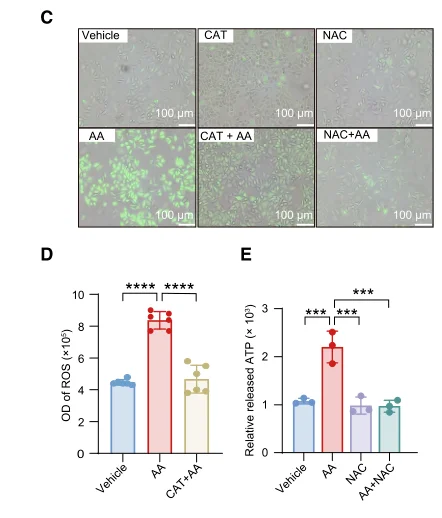
Lycopene (Cat. No. IL0510) serves as another reliable antioxidant agent for in vitro cell assays and in vivo animal tissue models, especially for hepatic injury and lipid peroxidation research. Comprehensive antioxidant efficacy evaluation requires multi-index detection across concentration gradients, solvent interference, ROS levels, endogenous antioxidant activity and cell viability; ignoring any of these readouts will lead to incomplete and one-sided mechanistic conclusions.
Before launching any oxidative stress detection assay, researchers must confirm reagent storage conditions, solvent solubility, batch quality certification and valid detection time windows. Experimental readouts are easily distorted by photodegradation, repeated freeze-thaw cycles and inconsistent cell lysis protocols. Information on setting up an assay or matching products can be found on the Solarbio technical services webpage or by browsing relevant technical articles.
Conclusion
Oxidative stress cannot be simply defined as excess ROS accumulation alone. It arises from a complex interactive network: unregulated ROS production overwhelms endogenous antioxidant clearance systems, leading to irreversible modification of DNA, proteins and membrane lipids. Oxidized lipid metabolites further amplify inflammatory cascades, which in turn aggravate oxidative damage and ultimately drive apoptotic cell death.
Cells deploy four core signaling axes to respond to redox imbalance: the Nrf2-ARE pathway initiates global antioxidant defense; NF-κB mediates oxidative stress-triggered inflammatory responses; MAPK cascades determine cell proliferation or apoptosis based on ROS dosage; the PI3K/Akt pathway maintains cell survival under mild redox perturbation.
Single-index detection cannot support rigorous oxidative stress research. To obtain comprehensive, reproducible data, simultaneous detection of multiple indicators is highly recommended, including total ROS concentration, antioxidant enzyme activity, MDA/4-HNE lipid peroxidation levels, phosphorylation status of key signaling proteins, pro-inflammatory cytokine secretion, mitochondrial function, apoptosis-related biomarkers and cell viability. If you need customized product matching, detailed experimental protocols or one-on-one technical support, feel free to reach out to Solarbio’s professional research service team.
FAQ
Q1: What exactly is oxidative stress?
A1: Oxidative stress refers to disrupted intracellular redox homeostasis, triggered by excessive ROS generation or impaired antioxidant clearance capacity. This imbalance induces biomolecular damage, aberrant signal transduction, inflammatory responses and apoptotic cell death, or a combination of these pathological phenotypes.
Q2: Is ROS inherently harmful to cells?
A2: No. Low-concentration, transient ROS act as essential physiological signaling messengers. Only sustained high ROS levels, or localized ROS overload within redox-sensitive organelles (mitochondria, nucleus), will exert cytotoxic effects.
Q3: Which core signaling pathway dominates cellular antioxidant defense?
A3: The Nrf2-ARE pathway serves as the master antioxidant regulatory axis. It transcriptionally upregulates SOD, CAT, GSH-Px, glutathione-dependent synthetic enzymes and phase II detoxification proteins to restore redox balance.
Q4: Why is NF-κB a vital research target for oxidative stress studies?
A4: NF-κB is the key molecular bridge connecting ROS overload and inflammation. It forms a self-amplifying positive feedback loop: ROS activates NF-κB to induce inflammation, and activated immune cells secrete extra ROS to further sustain pathway activation, worsening chronic tissue injury.
Q5: What key precautions should be taken when applying NAC in ROS experiments
A5: Researchers need to optimize NAC working concentration, incubation duration and solvent formulation in advance, and conduct parallel cell viability testing to exclude cytotoxic interference. Multi-index co-detection of redox biomarkers is required to generate solid, credible experimental conclusions.



