




Related news


HPLC LOD and LOQ Validation: How to Ensure Accurate and Compliant Results
Table of Contents
In HPLC method validation, LOD and LOQ are often written as two simple sensitivity values. In real laboratory work, they mean much more. These two parameters show whether an analytical method can detect or quantify low-level analytes with enough confidence, especially when the results will be used for quality control, method transfer, regulatory review, or routine sample testing.
Many validation problems do not come from the formula itself. They usually appear during sample preparation, baseline selection, matrix evaluation, or standard solution preparation. A method may show an acceptable signal-to-noise ratio in a clean solvent standard, but it may fail once real sample components, recovery loss, impurities, or baseline interference are introduced.
For this reason, LOD/LOQ validation should not only ask whether a peak can be seen. A better question is: Can this method detect and quantify the target analyte at low concentration in the actual sample matrix?
This article explains how to determine LOD and LOQ in HPLC, how to avoid common mistakes, and how to build a more reliable workflow for accurate chromatographic results.
What Are LOD and LOQ in HPLC?
LOD, or limit of detection, is the lowest concentration or amount of an analyte that can be detected under defined analytical conditions. It is mainly a qualitative parameter. LOD confirms that the analyte signal can be distinguished from background noise, but it does not prove that the analyte can be measured accurately.
In simple terms, LOD answers one question: Can the analyte be detected?
LOQ, or limit of quantitation, is the lowest concentration or amount of an analyte that can be quantitatively measured with acceptable accuracy and precision. Compared with LOD, LOQ has a higher requirement because it must support repeatable and reliable numerical results.
LOQ answers a stricter question: Can the analyte be accurately quantified?
The main difference is that LOD focuses on detectability, while LOQ focuses on quantitative reliability. In HPLC validation, this distinction is important because a visible peak is not always suitable for reporting a concentration.
LOD vs LOQ: Key Differences
| Parameter | LOD | LOQ |
|---|---|---|
| Full name | Limit of Detection | Limit of Quantitation |
| Main purpose | Confirms analyte presence | Measures analyte amount accurately |
| Typical S/N ratio | About 3:1 | About 10:1 |
| Data requirement | Detectable signal | Accuracy and precision |
| Validation focus | Sensitivity | Sensitivity, recovery, repeatability |
| Common use | Qualitative detection | Quantitative analysis |
Using the Signal-to-Noise Ratio Method in HPLC
In chromatographic analysis, the signal-to-noise ratio method is widely used to estimate LOD and LOQ during early method evaluation. In a typical HPLC workflow, analysts prepare diluted standard solutions at low concentration levels, run them through the HPLC system, review the chromatograms, and obtain the S/N value with chromatography software.

As a practical reference, LOD is usually associated with an S/N value of about 3:1, whereas LOQ is commonly linked to an S/N value of about 10:1.
The estimation can be written in the following form:
LOD = Prepared concentration × 3 / measured S/N
LOQ = Prepared concentration × 10 / measured S/N
During routine HPLC work, analysts often perform serial dilution until the target peak gives an S/N value near 3 for LOD or near 10 for LOQ. This value is useful for preliminary judgment, but it should not be regarded as final evidence that the method performs reliably at that concentration.
Why Baseline Selection Matters
A common source of error in LOD/LOQ determination is the use of an overly smooth baseline segment for noise measurement. When the chosen noise window shows very little fluctuation, the software may calculate an unrealistically low noise value. This can make the reported LOD or LOQ appear more sensitive than the actual method capability.
For reliable S/N calculation, the noise region should be close to the retention time of the target peak. It should be free from interfering peaks, but still representative of the real chromatographic baseline. A practical noise window is often at least five times the peak width, and in many cases 5–20 times the peak width is used.
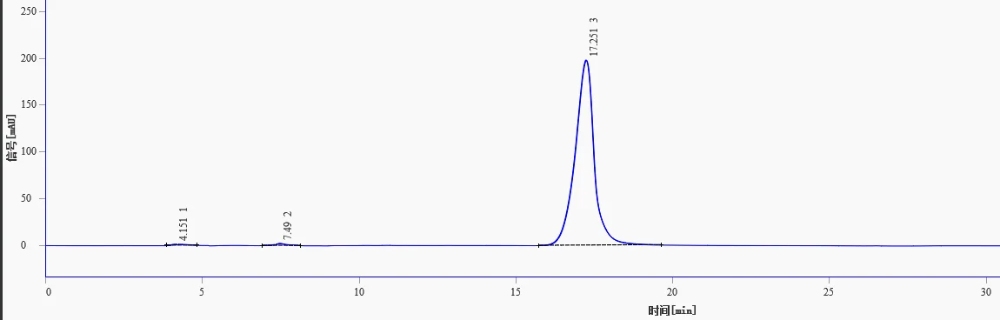
The key point is simple: do not choose a baseline only because it looks clean. The selected region should reflect the actual noise level around the analyte peak. Otherwise, the final LOD/LOQ values may look impressive on paper but fail during real sample analysis.
Why S/N Alone Is Not Enough for LOQ
A signal-to-noise ratio of 10:1 does not automatically prove that a concentration can be accepted as LOQ. LOQ must also meet accuracy and precision requirements. If a concentration gives S/N = 10 but has poor recovery or unstable replicate results, it should not be used as the final LOQ.
This issue often appears in HPLC method validation because real samples usually contain more interfering components than solvent standards. Depending on the sample type, the matrix may include impurities, degradation products, endogenous components, or co-eluting substances. These components can change peak shape, shift retention time, increase baseline noise, or alter detector response.
For this reason, LOQ validation should not rely on S/N evaluation alone. It should also include matrix-matched recovery and precision verification to confirm that low-level quantitation is reliable in real samples.
How to Validate LOQ Accuracy in HPLC
Use Matrix-Matched Spiked Samples
Standard solutions prepared in pure solvent are useful for initial estimation, but they cannot fully represent real sample conditions. Matrix components may influence extraction efficiency, analyte stability, chromatographic separation, and detector response.
A more practical approach is to use a blank matrix confirmed to be free of the target analyte. The analytical standard is then added to the blank matrix at the proposed LOQ level. After that, the sample should go through the complete sample preparation procedure before HPLC analysis.
A typical workflow includes:
Select a suitable blank matrix.
Spike the target analyte at the proposed LOQ concentration.
Process the sample using the full preparation method.
Inject the prepared sample into the HPLC system.
Calculate recovery and RSD.
Common practical acceptance criteria include recovery within 80%–120% and RSD generally not higher than 10%. However, these values should not be copied mechanically. The final acceptance range should be defined before validation begins and should match the analyte, matrix, method purpose, and regulatory requirements.
Verify Precision at the LOQ Level
Repeatability shows whether the method gives comparable results under the same laboratory conditions. This usually means the same laboratory, analyst, instrument, day, sample type, and LOQ-level concentration.
At least six replicate measurements are commonly used to evaluate repeatability. If the RSD is too high at the proposed LOQ level, the LOQ should be increased and verified again until the precision becomes acceptable.
Intermediate precision may also be evaluated when needed. This test checks whether the method remains reliable when conditions change, such as different days, analysts, or instruments. Although intermediate precision may allow slightly wider variation than repeatability, the method must still prove that low-level quantitation is stable enough for its intended use.
Control analytical standard Quality
The analytical standard has a direct effect on LOD and LOQ validation. If the standard is impure, unstable, incorrectly weighed, poorly dissolved, or degraded, the validation result will not be reliable.
For HPLC LOD/LOQ work, analytical standards should generally have suitable purity, preferably ≥98% when appropriate for the application. They should be within the validity period, stored under recommended conditions, and accurately weighed using a suitable analytical balance. The standard should also be completely dissolved in a compatible solvent.
For compounds sensitive to light, heat, oxidation, or repeated freeze-thaw cycles, small aliquots are recommended. This reduces degradation risk and improves concentration consistency during validation.
Poor-quality standards can lead to unstable calibration curves, inaccurate recovery, false sensitivity claims, and non-reproducible LOQ results.
IDL vs MDL: Do Not Confuse the Two
Instrument detection limit, or IDL, reflects the detection capability of the instrument itself. It is usually determined by directly injecting diluted standard solutions. IDL mainly answers: How sensitive is the instrument under ideal conditions?
Method detection limit, or MDL, includes the complete analytical process. It considers sample extraction, cleanup, dilution, matrix interference, instrument response, and data processing. MDL is usually more relevant for real sample testing because it reflects the performance of the entire method rather than only the instrument.
Many laboratories report IDL because it is easier to obtain and often looks lower. However, IDL may not be achievable in actual samples. For practical HPLC validation, MDL is often the more meaningful value.
Common LOD/LOQ Validation Mistakes
Several mistakes appear repeatedly in HPLC validation.
The first is reporting only S/N values. S/N is useful, but LOQ must also be supported by recovery and precision data.
The second is using solvent standards instead of matrix samples. Solvent standards may give cleaner baselines and better peak shapes than real samples, which can overestimate method sensitivity.
The third is selecting an unrealistically quiet baseline region. This can make calculated LOD and LOQ values appear lower than they truly are.
The fourth is ignoring standard stability. Degraded standards or repeatedly frozen-thawed solutions can cause incorrect concentration assignment.
The fifth is confusing IDL with MDL. Instrument capability does not equal full method capability.
Practical Workflow for HPLC LOD/LOQ Validation
A reliable workflow may follow this order:
Prepare a high-quality analytical standard solution.
Prepare serial dilutions near the expected low concentration range.
Inject each level and evaluate peak shape and S/N.
Estimate LOD at approximately S/N = 3.
Estimate LOQ at approximately S/N = 10.
Prepare matrix-matched samples at the proposed LOQ level.
Analyze at least six replicates.
Calculate recovery and RSD.
Confirm whether accuracy and precision meet predefined criteria.
If the criteria fail, raise the LOQ concentration and repeat verification.
This workflow is not complicated, but it helps prevent a common problem: accepting a low concentration too early simply because the chromatogram shows a visible peak.
How Analytical Standards Support Reliable HPLC Validation
Reliable analytical standards are essential for accurate LOD/LOQ determination. In HPLC method development, standards support calibration, recovery testing, system suitability, and sensitivity verification.

Solarbio provides analytical standards, standard solutions, stable isotope-labeled standards, and related technical support for chromatographic analysis. For research and testing laboratories, high-purity standards with traceable quality documentation can help reduce uncertainty in method validation.
Solarbio analytical standards are supported by multi-dimensional quality evaluation methods such as HPLC, NMR, and MS. The company also provides technical support for standard selection and HPLC testing services for complex samples. These resources are useful when laboratories need more confidence in low-level chromatographic data.
Why Choose Solarbio Analytical Standards?
Solarbio offers a broad selection of analytical standards and continues to develop products according to changing laboratory needs. Its standards are evaluated through multiple methods such as HPLC, NMR, and MS to support quality control.
For laboratories that need help with method setup, Solarbio can also provide support for standard selection, solvent choice, preparation guidance, and chromatographic condition optimization. For complex samples, HPLC testing services are available to help obtain more accurate and reliable data.
Solarbio’s testing center has obtained ISO/IEC 17025 recognition, supporting stronger analytical data quality and quality assurance.
Conclusion
LOD and LOQ validation in HPLC should not be reduced to a simple signal-to-noise calculation. A sound validation process should include proper baseline selection, S/N evaluation, matrix-matched recovery, precision verification, and analytical standard quality control.
LOD shows whether an analyte can be detected. LOQ proves whether the analyte can be quantified with acceptable accuracy and precision. For real sample analysis, method detection capability is more important than ideal instrument sensitivity.
By using suitable matrices, stable standards, and a complete validation workflow, laboratories can avoid common LOD/LOQ mistakes and generate chromatographic data that is more accurate, reproducible, and easier to defend during review. For more chromatography validation insights and laboratory workflow updates, visit Solarbio’s technical articles, or contact Solarbio for product selection and testing support.
FAQ
Q1: Why should LOQ be verified with matrix-matched samples?
A1: Real sample matrices can affect extraction efficiency, peak shape, baseline noise, and detector response. Matrix-matched samples make LOQ validation closer to actual testing conditions.
Q2: What recovery range is commonly acceptable at the LOQ level?
A2: A practical recovery range is often 80%–120%, but the final criteria should be defined according to the analyte, matrix, method purpose, and regulatory requirements.
Q3: How many replicate measurements are recommended for LOQ precision?
A3: At least six replicate measurements at the LOQ level are commonly used to evaluate repeatability and calculate RSD.
Q4: What should I do if the RSD at LOQ is too high?
A4: The proposed LOQ concentration should be increased and verified again until the precision meets predefined acceptance criteria.



